“我可以一天学完python,但是我没办法一天学完R”
前言
想入门生物信息的话,一定得做实战一篇文献的数据嘛,网上的教程林林总总,但完整从头到尾的实战少之又少,而且很多教程完全不是“入门”,只是拿着别人的代码复制粘贴,我在查资料的时候看的云里雾里的,正好自己在从零开始RNA-seq数据的分析,我把代码和流程写一份手册分享出来,希望对大家有一点启发,本人很菜,如果大家有什么评论或者建议的话欢迎去邮箱留言
本文所用的环境是windows 10 + Ubuntu系统
共勉
太川 2021/11/01 于大阪大学
本文可以随意转载和复制,但请注明出处!谢谢!祝一切顺利!
作者邮箱:huyajie@protein.osaka-u.ac.jp
wsl2
这是一个基于Windows系统的Linux虚拟机,微软自带的,之前我有使用过VMware,赶快卸载了用这个,太方便了!
我本来想写一个安装流程的,但是抱歉我忘记了当初怎么安装的,就是照着官方手册复制粘贴,好像遇到了一些小bug但google一下也解决了
也算是一个入门小考验吧(绝对不是博主懒得卸载重装一遍)
只是为了自学生物信息,发现bug解决问题从来都是我们的第一步,这是给大家一个锻炼的机会!
服务器
作为生物信息学分析,每一个数据都是几十G的,真要做个全分析一定要会连接和使用服务器,在这里放些如何连接服务器以及一些基础的Linux代码(cat,ls那种就不说了…,参考书籍:鸟哥的Linux私房菜)
连接服务器
这里我使用的是linux系统了
如果电脑使用的是Linux系统的话直接使用SSH就可
# 安装ssh
sudo apt install ssh
# 确认电脑上是否安装了客户端和服务器
dpkg -l|grep ssh
# 确认ssh-serve是否已经启动了
ps -e|grep ssh
# 连接服务器 ssh huiyoo@192.168.1.1
ssh username@IP
另外建议以windows操作的需要安装FileZilla,用于上传和下载服务器的文件到本地处理,例如在本地用Rstudio绘图,之后会用到
记住密码登录
如果不想每次输入密码的话看这里
# 在本地生成密钥
ssh-keygen -t rsa
# 将公钥复制到主机中
ssh-copy-id user@IP
# 直接用这个命令就可以登录了
ssh username@IP
常用的Linux指令
| 命令 | 作用 | 备注 |
|---|---|---|
| nohup | 不挂断程序 | 会生成nohup.out文件,cat查看进程 |
| & | 放在后台运行 | |
| ps | 查看瞬间进程的动态 | |
| jobs | 查看当前终端后台运行的任务 | |
| ls -lht | 查看文件大小 | |
| tar xvf | 解包.tar | tar cvf 打包.tar |
| gzip -d | 解压.gz | |
| tar zxvf | 解压.tar.gz和.tgz | tar zcvf 压缩 |
| jobs -l | 查看后台进程 | 想停止用kill [PID] |
| export LANG=en_US.UTF-8 | 把系统语言改成英语标码 |
Shell脚本
服务器进行工作的时候不可能一行一行完成了以后盯着打下一行,尤其是多数据分析和下载,这时候可以自己写一个脚本来自动运行,简单又方便
| 命令 | 作用 | 备注 |
|---|---|---|
| bash | 在子进程中执行 | 变量操作结束不会传回到父进程中 |
| source | 在父进程中执行 | |
| shift | 造成参数变量号码偏移 | |
| [ctrl]+u/k | 光标向前/向后删除指令 | |
| [ctrl]+a/e | 光标移动到命令串最前面/后面 | |
| unset | 取消设置的变量内容 |
使用qsub提交命令到服务器
因为一个服务器有很多人要同时使用,所以可以用qsub命令先提交指令,等待服务器安排多余的资源执行自己的指令,这就是qsub
# qsub脚本运行参考
# Run your jobs using the qsub commands.
qsub STAR.sh
# Run your jobs in the interactive mode.
qsub -I
# Display job status.
qstat
# Delete your job
qdel <job ID>
# STAR.sh
#!/bin/bash
########################################################################
#### -q option: Specify a type of your job, SMALL(1 cpu) or MEDIUM(2-8) or LARGE(9-64)
#### -V option: All environment variables are exported to the job.
#### -l option: Specify resources you want to use
########################################################################
#PBS -q MEDIUM
#PBS -l select=1:ncpus=8
#PBS -V
## move to the current directory
cd /home/username/DATA/RNA_seq_test/STAR_result
## your commands
echo "STAR start"
nohup STAR --runThreadN 8 --runMode genomeGenerate --genomeDir hg38_index_dir --genomeFastaFiles hg38.analysisSet.fa --sjdbGTFfile gencode.v38.annotation.gtf &
echo "STAR end"
环境配置conda及其他软件的下载安装
conda的重要性不用多说了,简单来说相当于一个生物信息软件的软件商店,我们很多软件和工具都是基于这个包去下载和使用的
# 安装conda
wget https://repo.anaconda.com/archive/Anaconda3-2021.05-Linux-x86_64.sh
# 运行
bash Anaconda3-2021.05-Linux-x86_64.sh
# 查看安装是否完成
conda -V
# 更新
# 我这里安装的目录可能和你不一样,但你只要输入conda update就可以根据提示更新了
conda update --prefix /home/username/software/anaconda3 anaconda
# 配置环境(每个项目创建一个单独的环境,防止环境污染)
conda create -n BioInf python=3.8
# 查看当前环境
conda info -e
# 激活环境
conda activate BioInf
# 添加镜像配置环境(bioconda)
conda config --add channels conda-forge
conda config --add channels r
conda config --add channels bioconda
# 由于我在国外,国内的需要配置清华镜像,google以后有很多,可以参考一下这篇文章,博主自己没有试,因为会有更新种种问题
https://www.cpci.dev/anaconda-mirrors-configure/
# 查看配置的镜像
conda config --get channels
# 安装软件
conda install ..
# 软件列表
conda install Samtools
conda install Bedtools
conda install star
conda install fastqc
conda install sra-tools
conda install trim-galore
conda install seqkit
conda install Subread
# 这里可能存在版本问题导致fasterq-dump无法使用,更新一下就好了
conda update sra-tools
# 全部更新,强迫症最喜欢的一条命令
conda update --all
原始数据下载
开头我想强调一下,本文的代码有一个明显的Bug,就是我省略了新建文件夹和移动的步骤,因为每个人的保存习惯都不一样,命名方式也不一样,所以如果只是复制粘贴是没办法运行的,请根据自己的习惯修改成自己的路径再运行
我希望看到这篇文章的大家是抱着学习的态度来的,一定看清楚我每个参数为什么这么设置,复制粘贴什么都学不会,所以故意设置这个bug是为了让大家写属于自己的代码
这里测试使用的参考文献是这篇,流程是助教提供的
https://www.nature.com/articles/ng.3844
以SRR2183367,SRR2183369,SRR2183371,SRR2183373数据为例
这里先说明一下免得看下去比较迷糊,两种细胞,每种细胞做了一个重复,建议把所有数据都下载下来,我们在最后要一起做个比较,否则至少下两种细胞的数据,例如PN1和CL1
| 名字 | ID | 细胞种类 |
|---|---|---|
| PN1 | SRR2183367 | Pronuclear stage(1-cell) |
| PN2 | SRR2183369 | Pronuclear stage(1-cell) |
| CL1 | SRR2183371 | Cleavage(2-,4-,8-cell) |
| CL2 | SRR2183373 | Cleavage(2-,4-,8-cell) |
搜索下载源
fasterq-dump
fasterq-dump自带了下载和解压工具,只要一行代码,是比较好用的下载方法,另外我也找到了其他的下载方法放在下面,但这里我不清楚自动下载的目录和文件夹,因为使用下文提到的prefetch命令会默认创建一个/ncbi/public/sra文件夹,文件都默认保存在那里
20211104补充:不会新建文件夹,自动下载在命令运行时的目录下
另外fasterq-dump通俗点来讲是个解压或者翻译软件?利用Aspera或者prefetch下载下来的.sra文件是二进制编码,将它变成可读fastq文件就需要fasterq-dump了
总结来说能用fasterq-dump一行代码可以下载和编译就很方便了
# SRR_ID for example SRR2183367
nohup fasterq-dump --threads n --progress SRR_ID &
# --threads 使用核心数量n --progress显示进程
nohup fasterq-dump --progress SRR2183367 &
nohup fasterq-dump --progress SRR2183371 &
# 下载两个是因为之后会需要两个数据作比对,其实最好全下了,我们之后绘图会用到
在这里解释一下为什么下载之后是两个文件_1,和_2。是因为我们用的数据是双端测序数据,如果只有一个文件则说明是单端测序,双端测序简单来说就是测序是分别从两端向中间测序,保证测序的准确性,1和2文件就是两端
20211215修订:作者在做这套流程的时候还不会shell脚本,虽然现在会了,但我还是把第一次写的代码放上来了,至于脚本怎么写具体可以参考文末的老师代码学习
Aspera Connect
Aspera是一种下载速度较快的下载方式,如果不行可以用SRA toolkit的prefetch命令
安装
wget http://download.asperasoft.com/download/sw/connect/3.7.4/aspera-connect-3.7.4.147727-linux-64.tar.gz
# 解压缩
tar zxvf aspera-connect-3.7.4.147727-linux-64.tar.gz
# install
bash aspera-connect-3.7.4.147727-linux-64.sh
# check the .aspera directory
cd # go to root directory
ls -a # if you could see .aspera, the installation is OK
# add environment variable
echo 'export PATH=~/.aspera/connect/bin:$PATH' >> ~/.bashrc
source ~/.bashrc
# 密钥备份到/home/的家目录(后面会用,否则报错)
cp ~/.aspera/connect/etc/asperaweb_id_dsa.openssh ~/
# check help file
ascp --help
下载格式
ascp -v -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh -k 1 -T -l200m anonftp@ftp-private.ncbi.nlm.nih.gov:/sra/sra-instant/reads/ByRun/sra/SRR/SRR949/SRR949627/SRR949627.sra .
SRA toolkit的prefetch命令
下载单个SRA文件
如果安装了Aspera,prefetch会优先使用ascp协议下载
prefetch SRR2183367
下载多个SRA文件
例如,点击accession list,将下载得到的文本文件上传到服务器/home/Seqs目录,关于服务器的上传可以使用File Zilla
# 运行命令
prefetch --option-file Seqs/SRR_lists.txt
质量控制
这里为了便于理解不同工具处理数据的不同,我建议大家每做完一个步骤可以测一个FastQC看看到底哪里有不同,我在实际操作的时候在FastQC_result文件夹下面新建了例如1_FastQC,2_FastQC等文件夹分别放入结果
# 下载后生成两个fastq文件,这里是同一阶段的两个复制
SRR2183367_1.fastq SRR2183367_2.fastq
# 读取前100万个reads构建小型数据作为练习,并命名为PN1_1和PN1_2,因为4行为一个read所以要*4
head -4000000 SRR2183367_1.fastq > PN1_1.fastq
head -4000000 SRR2183367_2.fastq > PN1_2.fastq
# fastQC检查质量
# 创建一个文件夹保存结果,-p如果该文件夹存在就创建,不存在就保留原文件夹
mkdir -p ./FastQC_result
# -t调用两个核 -o输出到哪个文件 -q沉默模式
fastqc -t 2 -o ./FastQC_result/ -q *.fastq &
# 这里是想查看本地电脑中linux的文件,根目录在
\\wsl$\Ubuntu\home\username
# 或者可以在Linux系统想打开的目录中输入
explorer.exe .
对于FastQC的报告解读,网上有很多详细的说明,我看的是这篇文章
PS:我觉得这是一个好习惯,包括你每次处理数据的时候把不同的数据整理到不同的文件夹里
# 例如这样
11月 10 16:40 STAR_result
11月 10 15:38 seqkit_result
11月 10 15:18 FastQC_result
11月 10 15:18 rRNAdust_result
11月 10 13:54 trim_report
11月 10 12:27 origin_data
前处理
去除adapter和低质量碱基
这一步是因为在第二代测序的时候,我们会使用引物去复制测序,这一步是切掉引物,fastqc报告可以很显著的看出这一步的结果
# 使用trim_galore去除低质量碱基和adapter,并输出到trim_report文件夹中
trim_galore -q 20 --nextera --paired *.fastq --gzip -o trim_report &
# --nextera 使用的是这个的adapter,根据自己的测序数据需要变换引物,貌似也可以不加自己识别
查看去除引物之后的序列长度
这一步需要使用正则表达式处理数据并输出文件,训练使用R语言绘图查看信息
正则表达式是一种很好用的文本处理工具,例如awk,sed等,我看的是这个视频
做到这里你可能有点挫败感,什么要学的东西居然有这么多,那肯定的,慢慢来吧,每天都在进步
# 不做这一步也没关系,只是正则表达式的练习,脚本的编程以及R语言的使用
# 下面这一长串简单来说就是读取所有.fq为后缀的文件,计算基因片段的长度,小于70的归类为'<70',比较不同样品各个片段的长度
for file in *.fq; do name=$(echo $file | cut -f1 -d"_"); cat $file | awk -v n=$name 'NR%4==2{len=length($0);if(len<70) len=70;h[len]+=1} END{OFS="\t";for(key in h) print key,h[key],n}'; done | sort -k1,1n -k3,3 | sed 's/^70/<70/g' > dist.txt
这一步开始在本地查看结果导入RStudio绘图
关于R语言的学习和绘图,作者使用的参考书是R语言实战,结合网上北大李老师自制的学习笔记
# 从这里开始在RStudio文件开始绘图
# 再强调一遍,注意我的路径和你的不一样
# 读取数据
> dist <- read.table('C:/Lab/DATA/dist.txt',fill=TRUE)
# 将偶数行的数据和奇数行的合并,就是提取偶数行的数据和奇数行的加起来输出到奇数行
# 关于这一步作者想了很多方法,其实最简单的应该是在上一步使用正则表达式就可以优化一下,总之现在回头看看这方法真蠢,但是能运行我就不修改了
> dist[seq(1,nrow(dist),2),2] <- dist[seq(1,nrow(dist),2),2] + dist[seq(0,nrow(dist),2),2]
# 删除偶数行的数据
> dist <- dist[-seq(0,nrow(dist),2),]
# 设置因子
> bp <- factor(dist$V1,levels = c('<70',71:101))
# 绘图
> p <- ggplot(dist, aes(x=bp,y=V2,color=V3)) + geom_col()
# 小小的优化一下
> p <- ggplot(dist, aes(x=bp,y=V2,fill=V3)) + geom_col(width = 0.5) + xlab(NULL) + ylab(NULL) + theme(legend.title = element_blank());p
# legend.title = element_blank() 移除图例标题
# 关于数据堆叠,这是由于geom_col()中有一个默认参数position = 'stack'导致的
## 在针对X轴或者Y轴排序问题的时候可以使用forcats()是一个很好的分类变量处理工具
最后的图片是这个样子
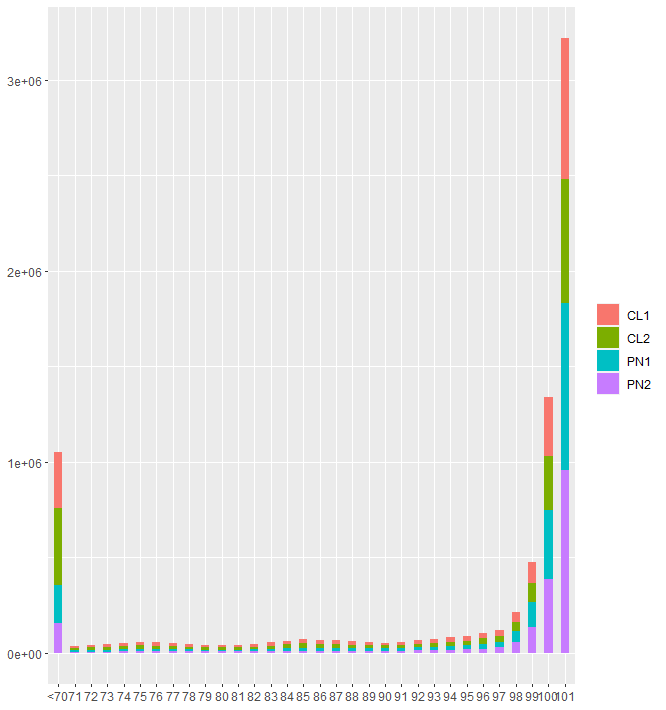
rRNAdust
安装
# 这里rRNAdust的作用是除去冗杂的rRNA,貌似这个测序Helicos数据集的错误率很高,所以要把错误丢弃
# 安装rRNAdust
wget https://fantom.gsc.riken.jp/5/suppl/rRNAdust/rRNAdust1.06.tgz
tar -zxvf rRNAdust1.06.gz
# 这里首先如果第一次的话要安装Make和gcc进行编译
sudo apt install make
sudo apt install gcc
# 在编译的时候有一些小Bug,系统提示先升级一下
sudo apt-get update
# cd到解压文件夹目录
make
# 这时候输入rRNAdust提示无法运行,因为还没有把它放入bin文件夹中
sudo cp rRNAdust /usr/bin
# 这里用实验室服务器的话会遇到一个bug,提示无权限,你只要在自己的文件夹下新建一个bin然后导入PATH就好了
mkdir $HOME/bin
export PATH=$PATH:$HOME/bin
cp rRNAdust $HOME/bin
除去冗杂的rRNA
下载rDNA sequence file,这里我这个笨比还不知道如何用linux下载,所以我用的复制粘贴
https://www.ncbi.nlm.nih.gov/nuccore/U13369.1?report=fasta
# 使用rRNAdust处理输出
cat PN_1_val_1.fq | rRNAdust -t 8 U13369.1.fa > out_PN1_1.fq
cat PN_2_val_2.fq | rRNAdust -t 8 U13369.1.fa > out_PN1_2.fq
拼接两个fastq文件
用拼接是翻译问题,或者说修复还是配对可能更好一点
# 使用seqkit比较两个fastq文件,这里因为使用rRNAdust把污染的部分去除了,因为这个数据是双端测序,所以去除了一端,另一端也要去除
seqkit pair -1 out_PN1_1.fq -2 out_PN1_2.fq -o result
比对
生成基因组索引文件
# 下载Gencode annotation,他是基因组标准注释文件数据库,下载main即可(GTF格式)
wget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_38/gencode.v38.annotation.gtf.gz
# 下载参考基因组序列(FASTA文件)
wget https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/analysisSet/hg38.analysisSet.fa.gz
# 解压
gunzip gencode.v38.annotation.gtf.gz
gunzip gencode.v38.annotation.gtf.gz
# 构建基因组索引,这里的hg38_index_dir需要提前建立
mkdir hg38_index_dir
# 这里提醒一下,这一步需要消耗很多内存,所以如果是学校服务器的话在前文提到的用qsub命令,shell已经给你写好了,我在学校服务器上跑的大概二十分钟
# 这个步骤只要做一次就好了,索引文件就相当于字典的目录
nohup STAR --runThreadN 8 --runMode genomeGenerate --genomeSAsparseD 3 --genomeSAindexNbases 12 --genomeDir hg38_index_dir --genomeFastaFiles hg38.analysisSet.fa --sjdbGTFfile gencode.v38.annotation.gtf &
这里再安利一个翻译软件deepl,女朋友推荐的!好用!
Mapping
这一步是为了将我们测得和处理过的fq文件回帖到基因组中的位置,就是要根据字典的目录查每个基因是什么
关于BAM文件的信息可以参考这篇文章,也许会对生成数据索引有新的理解
# 其中--outFileNamePrefix指的是新建的文件前缀,每个都不一样记得改
STAR --genomeDir hg38_index_dir --runThreadN 24 --readFilesIn out_PN1_1.paired.fq out_PN1_2.paired.fq --outFileNamePrefix PN1_ --outSAMtype BAM Unsorted SortedByCoordinate --quantMode GeneCounts
# 这里我遇到个bug
# bug信息
BAMoutput.cpp:27:BAMoutput: exiting because of *OUTPUT FILE* error: could not create output file
# 查了文档,用这行代码解决了,但不知道为什么
ulimit -n 10000
计算表达量
# 使用featureCount计算表达量
# 这一步可以把所有数据生成到一个txt文件中
# 注意文件路径
featureCounts -T 6 -p -t exon -g gene_id -a /home/HuYajie/DATA/RNA_seq_test/STAR_result/gencode.v38.annotation.gtf -o Counts.txt *.bam
利用edgeR进行差异化分析
这里就不在Linux里操作了,把Counts.txt文件下载下来,我们换RStudio用edgeR包做差异化分析
安装R包
# 首先需要安装BiocManager
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(version = "3.14")
# 安装edgeR
BiocManager::install('edgeR')
# 启用包
library(edgeR)
R代码运行部分
# 数据导入,注意这里需要更改自己的文件路径
> mydata <- read.table("C:/Lab/DATA/RNA_seq_test/Counts.txt",header = TRUE, quote = '\t', skip = 1)
# header = TRUE 第一行为变量名的逻辑变量,quote 用于指定包围字符型数据的字符,skip 读取数据时忽略的行数
# 更改分组名称
> sampleNames <- c("CL1","CL2","PN1","PN2")
> names(mydata)[7:10] <- sampleNames
# 使用数据的后面四列样本来组成矩阵
> countMatrix <- as.matrix(mydata[7:10])
> rownames(countMatrix) <- mydata$Geneid
# 创建DEGlist
> group <- factor(c("CL","CL","PN","PN"))
> y <- DGEList(counts = countMatrix,group = group)
# DEGList是一个可以包含多种内容和统计的列表。DGEList至少需要的元素:counts、samples(包含group分组信息和lib.size文库大小),counts用来存放表达矩阵,samples用来标记样本信息和库的大小,group声明组别
# 过滤掉为0的数据
# CPM(counts per million),此列设置最小组内样本数为2
> keep <- rowSums(cpm(y)>1) >= 2
> y <- y[keep,,keep.lib.sizes=FALSE]
# 标准化处理
> y <- calcNormFactors(y)
# 建立设计矩阵,为了更好的分组分析数据
# 这一步的意思是提取了CL1,CL2,PN1,PN2字符串的数字部分作为因子输出,作为一个分组的参考,分组其实根据数据决定,后面经老师修改这一步其实多余了
# subGroup <- factor(substring(colnames(countMatrix),3,3))
> design <- model.matrix(~group,y)
# 评估离散度
> install.packages('statmod')
> y <- estimateDisp(y,design,robust = TRUE)
# 查看离散度
> y$common.dispersion
> plotBCV(y)
# 估算每个基因的负二项离散Tagwise,(即经验贝叶斯稳健离散值)、Common(即经验贝叶斯稳健离散值的均值)、Trended(即经验贝叶斯稳健离散值的拟合值),robust=TRUE 代表稳健性线性回归
# 构建广义的线性模型(这里其实我也不太懂,其实这部分都需要补一下概率论的知识,但我们先硬着头皮做完,再回头补补课吧)
> fit = glmFit(y, design)
> lrt = glmLRT(fit,coef=2)
> topTags(qlf)
# 输出结果 topTags 是指提取出表达量最不相同的基因,基于'fdr'
> results = topTags(lrt, adjust.method="fdr", n=dim(y)[1])$table
# 将基因名赋值给key
> key = rownames(results)
# 有些基因的表达量很高,我们log一下
> logcpm = cpm(y, log=TRUE)
# 输出结果
> results = cbind(logcpm[key,], results$logFC, results$logCPM, FDR=results$FDR)
# 准备作图
# 这里我老师把这个表输出了一个文件,我们节约一点时间
> result <- results
# 表格是matrix,我们转换成data.frame方便作图
> result <- data.frame(result)
# PN1-PN2
> p <- ggplot(result,aes(x=PN1,y=PN2)) + geom_point();p
# PN1-CL1
> p <- ggplot(result,aes(x=PN1,y=CL1)) + geom_point();p
# CL1-CL2
> p <- ggplot(result,aes(x=CL1,y=CL2)) + geom_point();p
# 完美结束,恭喜你!
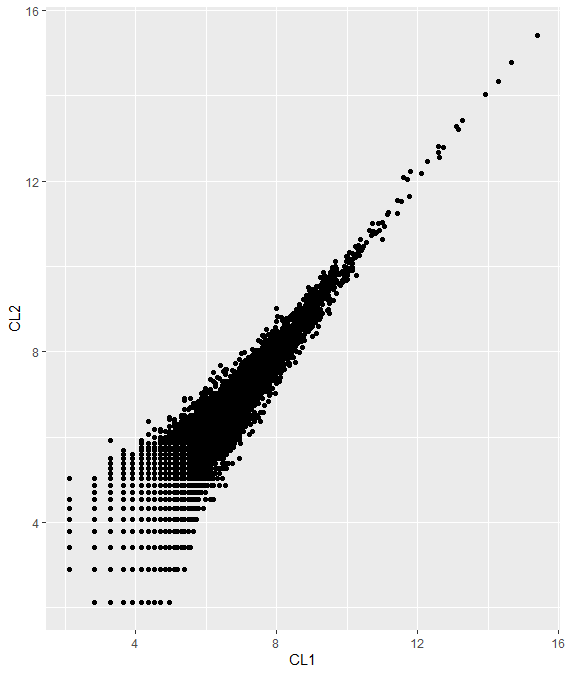
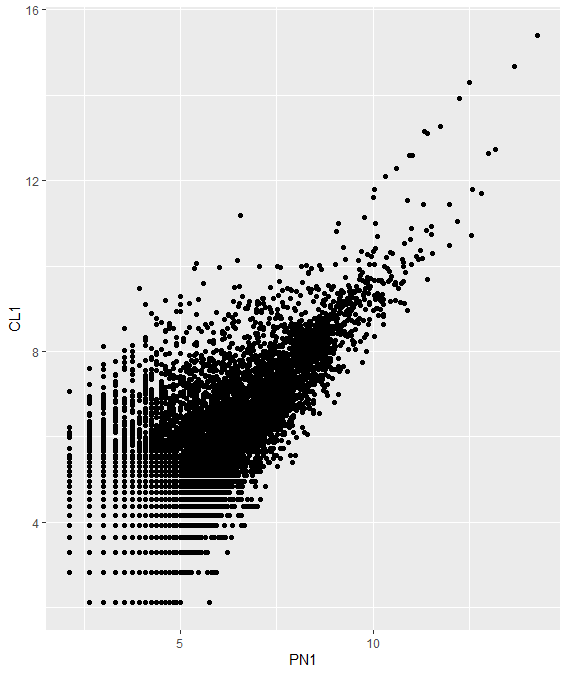
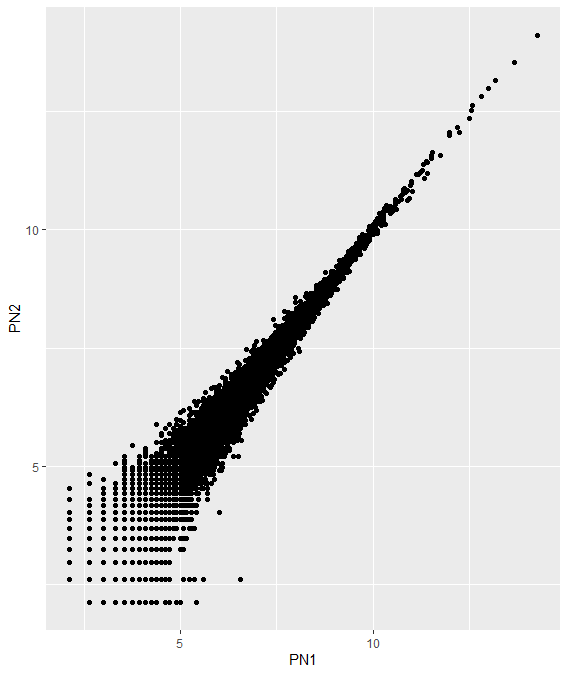
老师代码参考
# shell
Examples of commands
#### 3. Extract 1 million reads from each fastq file, and generate a small dataset for testing
mkdir test
cd test
for file in ../*.[12].fq; do
name=$(basename $file) && echo $name
cat $file | head -4000000 > $name
done
### 5. Remove nextera adapter sequences using trim_galore with "--nextera" option
for file in 0*.1.fq; do
name=$(basename $file .1.fq)
trim_galore --paired --nextera ${name}.1.fq ${name}.2.fq
done
### 5.1 Look at the size distribution after removing the adapter
for file in 0*val_[12].fq; do
name=$(echo $file | cut -f1-3 -d".")
cat $file | awk -v n=$name 'NR%4==2{len=length($0); if(len<70) len=70; h[len]+=1}
END{OFS="\t"; for(key in h) print key, h[key], n}'
done | sort -k1,1n -k3,3 | sed 's/^70/<70/' > 61.dist.txt
### 11. Generate human genome index files using "STAR --runMode genomeGenerate"
STAR --runMode genomeGenerate --runThreadN 8 --sjdbGTFfile hg38.gencodeV36.gtf \
--genomeDir hg38 --genomeFastaFiles hg38.fa
### 12. Run mapping through qsub using STAR
qsub -v name="PN1" qsub.STAR.sh
---- qsub.STAR.sh ----
#!/bin/bash
#PBS -q MEDIUM
#PBS -l select=1:ncpus=8
#PBS -V
cd $PBS_O_WORKDIR
fa="$GENOME/STAR/hg38"
cpu=8
STAR \
--readFilesIn ${name}.1.fq.gz ${name}.2.fq.gz \
--runThreadN $cpu \
--genomeDir $fa \
--outSAMunmapped Within \
--outSAMtype BAM SortedByCoordinate \
--readFilesCommand zcat \
--outFileNamePrefix 99.mapping/$name
mv 99.mapping/${name}Aligned.sortedByCoord.out.bam 99.mapping/${name}.bam
samtools index -@ $cpu 99.mapping/${name}.bam
rm -rf 99.mapping/${name}Log.progress.out 99.mapping/${name}Log.out 99.mapping/${name}_STARtmp
---------------------------
# Rstudio
library(edgeR)
## read the count file generated by featureCounts (need to remove headers)
tbl = read.table("22.count.txt", row.names=1)
grp = factor(c(1,1,2,2))
dge = DGEList(counts=tbl, group=grp)
dge = calcNormFactors(dge)
dsn = model.matrix(~grp)
dge = estimateDisp(dge, dsn)
## To perform likelihood ratio tests:
fit = glmFit(dge, dsn)
lrt = glmLRT(fit,coef=2)
results = topTags(lrt, adjust.method="fdr", n=dim(dge)[1])$table
key=rownames(results)
logcpm = cpm(dge, log=TRUE)
results = cbind(logcpm[key,], results$logFC, results$logCPM, FDR=results$FDR)
write.table(results, "23.count.cpm.txt", quote=FALSE, sep="\t", col.names=FALSE, row.names=TRUE)
草稿
为什么要留草稿呢,因为里面好多东西万一以后用的上呢
# 保存双端测序
fasterq-dump -I --gzip --split-files SRR218367.sra &
-I # 增加编号
--zip # 重新压缩
--split-files # 解压双端文件
& # 后台运行
# less命令打开检查
# 创建一个文件夹,保留结果
mkdir -p ./FastQC_result # -p如果该文件夹存在就创建,不存在就保留原文件夹
# -t调用两个核 -o输出到哪个文件 -q沉默模式
fastqc -t 2 -o ./FastQC_result/ -q ./raw.fastq/*.gz &
# -o 3 当有3bp的adapt序列能和read序列overlap上 -m 75 当序列有一条低于75的时候这一对都不要了 -a -A 分别对read1和read2取5'端的adapt -o输出内容
nohup cutadapt --times 1 -e 0.1 -o 3 --quality-cutoff 6 -m 75 -a AGATCGGAAGAGC -A AGATCGGAAGAGC -o $out_fastq_1 -p $out_fastq_2 $fastq_1 $fastq_2 > $log_file 2>&1 &
nohup tophat2 -p 32 -o $output_dir $mm10_index $fastq_1 $fastq_2 > $log 2>&1 &ss
# 这里遇到了Bug,当我使用STAR时提示内存不够,因为先用笔记本的Ubuntu系统只有3G的内存,需要新建配置文件,注意这里不是新建txt文件,而是要用pycharm等软件新建,或者用Linux自带的vim也可以,然后打开Powershell关闭wsl(wsl --shutdown),重启即可
新建C:\Users\\<yourUserName>\\.wslconfig
修改为如下内容(不要分割线):
--------------------------------------
[wsl2]
memory=32GB
processors=12
---------------------------------------
# 构建索引之前先用seqkit把fq文件抓换成fa文件
seqkit fq2fa out_PN1_1.paired.fq -o PN1_1.fa
# 这里做个分割线,其实后面的部分不需要
> result <- y$counts
> result <- data.frame(result)
# 限定坐标轴
> p <- ggplot(result,aes(x=PN1,y=PN2)) + geom_point() + ylim(0,15) + xlim(0,15);p
# 查看差异表达基因原始的CMP(就是原始表达量)
> top <- rownames(topTags(qlf))
> cpm(y)[top,]
# 查看上调和下调的基因的数目
> summary(dt <- decideTestsDGE(qlf))
# 挑选出差异表达基因的名字
> isDE <- as.logical(dt)
> DEnames <- rownames(y)[isDE]
> head(DEnames)
[1] "ENSG00000228463.10" "ENSG00000225630.1"
[3] "ENSG00000248527.1" "ENSG00000228794.10"
[5] "ENSG00000234711.1" "ENSG00000188976.11"
# 差异表达基因画图
> plotSmear(qlf,de.tags = DEnames)
> abline(h=c(-1,1),col="blue")
在写手册的时候突然想到,如果把生物信息的流程写成一套程序挂在steam上,加上成就系统和游戏时间,这样学起来会不会更有乐趣一点,然后把不同的数据库写成不同的DLC,我可真是天才(但我目前写不出来就是了)